Corticothérapie
Les glucocorticoïdes (GC) (ou “corticoïdes”) de synthèse sont des médicaments qui dérivent de l’hormone naturelle issue de la corticosurrénale, le cortisol. Ils furent développés pour maximiser les effets glucocorticoïdes, en particulier anti-inflammatoire, tout en minimisant les effets minéralocorticoïdes, tels que anti-diurétique, anti-natriurétique et kaliurétique.
Physiologie
Cortico-surrénale
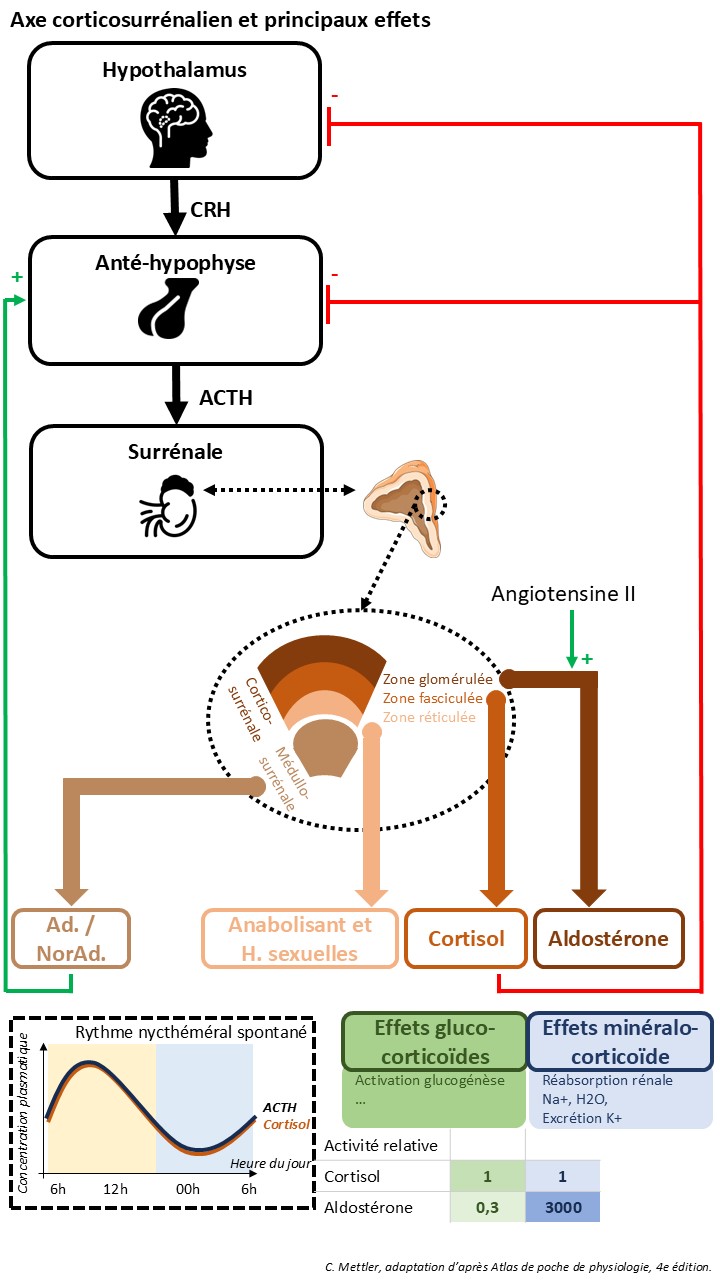
Le cortex surrénalien (ou corticosurrénale (CS)) produit dans le zone fasciculée les glucocorticoïdes, tandis que les minéralocorticoïdes (aldostérone, corticostérone et 11-désoxycorticostérone) et les androgènes de la CS sont synthétisés respectivement par la zone glomérulée et la zone réticulée. Les hormones stéroïdes sont d’un intérêt vital, en particulier pour l’adaptation aux situations de stress.
Le stimulus pour la sécrétion de cortisol est l’ACTH (hormone adrénocorticotrope), issue de l’hypophyse, et pour l’aldostérone principalement l’angiotensine II. La sécrétion d’ACTH est stimulée par la CRH (corticoréline ou corticotropin releasing factor), sécrétée par l’hypothalamus, et l’adrénaline, et inhibée par rétrocontrôle négatif par le cortisol (en partie par l’intermédiaire d’un rétrocontrôle sur le CRH également).
Il existe un rythme nycthéméral spontanée de la sécrétion de CRH, et par conséquent de l’ACTH et du cortisol, dont le maximum se situe le matin.
Biosynthèse des hormones stéroïdes
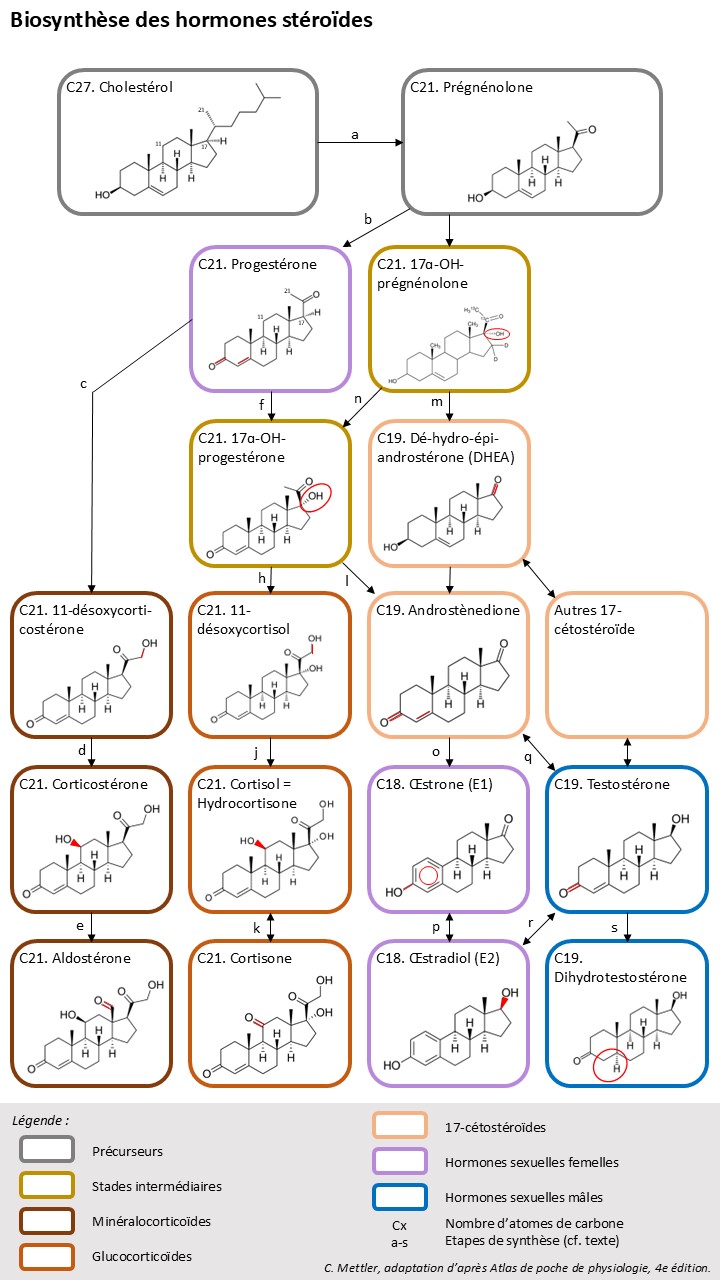
Le cholestérol est la substance de base des hormones stéroïdes. Il est synthétisé principalement dans le foie puis transporté dans les lipoprotéines vers les glandes endocrine. Il peut également être synthétisé de novo dans la CS. Les hormones stéroïdes sont stockées en faible quantité sur leur lieu de production (CS, ovaires, testicule, placenta), ainsi en cas de besoin elles doivent être synthétisées à partir de la réserve cellulaire de cholestérol.
Le cholestérol contient 27 atomes de carbone. Après plusieurs étapes intermédiaires, il est transformé en prégnénolone [a], contenant 21 atomes de carbone, précurseur des hormones stéroïdes. A partir de la prégnénolone est synthétisé la progestérone [b], hormone sexuelle femelle mais également point de départ de la synthèse de toures les autres hormones stéroïdes : les hormones de la CS avec 21 atomes de carbone, les hormones sexuelles mâles (androgène) avec 19 atomes de carbone (dans le testicule, l’ovaire et la CS) et les hormones sexuelles femelles (œstrogènes) avec 18 atomes de carbone.
Les substances de base de la synthèse des hormones stéroïdes sont présente dans toutes les glandes hormonales stéroïdiennes. Le type d’hormone et le lieu de production sont fixés par la présence de récepteurs aux hormones de contrôle de rang supérieur (ACTH, FSH, LH, …) et la prédominance d’un type d’enzyme intervenant sur la structure moléculaire des stéroïdes dans les cellules de la glande hormonale concernée. La CS contient les enzymes 17-, 21 et 11- hydroxylases (ajout d’un groupement OH sur l’atome de carbone correspondant).
Dans la zone glomérulée de la CS, l’hydroxylation de l’atome 21 [c] rend le stéroïde non disponible pour la 17-hydroxylase, ainsi seuls les minéralocorticoïdes (corticostérone, aldostérone) sont synthétisés. Si l’hydroxylation à lieu d’abord sur l’atome C17 [f et g], la voie de synthèse conduit aux GC (surtout dans la zone fasciculée de la CS [h-j-k]) et aux 17-cétostéroïdes (groupement cétone sur le C17 [l-m]). Les glucocorticoïdes et les 17-cétostéroïdes peuvent également être synthétisés en contournant la progestérone, partir de la 17ɑ-OH-prégnénolone [n-m]. Les 17-cétostéroïdes peuvent conduire aux œstrogènes (œstrone et œstradiol) par une voie directe [o-p] ou par une vie indirecte avec comme intermédiaire un androgène, la testostérone [q-r-p].
La dégradation des hormones stéroïdes se fait essentiellement dans le foie. Elles y sont conjuguées à des sulfates ou des acides glucuroniques, puis excrétées par la bile ou l’urine.
Effets physiologiques des hormones stéroïdes
Le cortisol stimule dans le foie la synthèse de glucose à partir des acides aminés (glucogénèse) et protège l’organise d’une hypoglycémie après un jeun prolongé lorsque les réserves hépatiques de glycogène sont épuisées. Il empêche également les réactions inflammatoires excessives.
L’aldostérone stimule la réabsorption de Na+, Cl- et H2O et lutte ainsi contre une réduction du volume du liquide extracellulaire.
Pharmacodynamie
Les GC ont un mécanisme d’action génomique lent (quelques heures à jours) et un mécanisme d’action non-génomiques rapide (quelques minutes)
Mécanismes d’actions
Mécanismes génomiques
Les GC agissent principalement via le récepteur aux glucocorticoïdes (RG), un récepteur nucléaire ubiquitaire, appartenant à la superfamille des récepteurs aux stéroïdes (récepteur en doigt de zinc). Ce récepteur est présent sous une forme inactive dans le cytosol, lié à un complexe protéique comprenant la " heat-shock protein " (HSP) 90 et l’immunophiline. Les GC diffusent passivement à travers la membrane cellulaire puis se lient au RG, provoquant la dissociation de ce complexe protéique.
Le complexe GC/RG, sous forme d’homodimère, transloque vers le noyau, où il module l’expression de gènes cibles par trois mécanismes principaux :
- Action transcriptionnelle directe : liaison d’un homodimère de RG à une séquence ADN appelée Glucocorticoid Response Element (GRE), activatrice de la transcription. Cette liaison permet une augmentation de production de protéines anti-inflammatoires comme la lipocortine-1 (ou annexine-1), l’interleukine 10 ou la protéine IkB. Ce mécanisme de transactivation est responsable d’une partie des effets thérapeutiques, mais aussi de certains effets indésirables métaboliques (ex. : hyperglycémie, ostéoporose).
- Action transcriptionnelle indirecte : intéraction de type protéine-protéine entre le RG et des facteurs de transcription NF-kappa B, NF-IL6, AP-1 et STATS conduisant à une inhibition de ces facteurs et donc à une transrépression des gènes cibles (Cytokines IL-1, IL-2, IL-6, IL-12, TNF-ɑ, IFN-ɣ ; chimiokines MCP-1, IL-8, eotaxine et enzymes inflammatoires COX-2, …). Ce mécanisme est central dans l’effet anti-inflammatoire et immunosuppresseur des GC et semble moins associé aux effets indésirables.
- Action sur la structure chromosomique : modification de la structure de la chromatine, réduisant l’accès des facteurs de transcription à leurs sites de fixation et inhibant l’expression des gènes concernés.
Mécanismes non-génomiques
Les GC ont également des mécanismes d’action non génomiques, telles que les intéractions membranaires ou des modifications post-transcriptionnelles, permettant des effets plus rapides, comme la vasoconstriction ou l’inhibition de la dégranulation des mastocytes. Indépendants de la transcription et de la traduction, ces mécanismes sont importants à fortes doses, comme lors de la thérapie par bolus ( pulse therapy ).
NB : Dose-Dépendance et Saturation du RG : La saturation du RG est presque complète aux doses élevées (30–100 mg d'équivalent prednisolone par jour), ce qui est utilisé dans les situations d'urgence ou dans les maladies auto-immunes sévères. La thérapie par bolus (>250 mg) est supposée exploiter les mécanismes non-génomiques.
Effets pharmacodynamiques des GC en clinique
- Effets anti-inflammatoires :
Inhibition de la production de cytokines pro-inflammatoires (IL-1, IL-6, IL-8, TNFalpha) -> Diminution de l’afflux de macrophages et de granulocytes sur le site inflammatoire.
Inhibition de l’expression de molécules d’adhésion (ICAM) -> Diminution de la migration transendothéliale des cellules phagocytaires.
Inhibition de la phospholipase A2 et de la cyclooxygénase de type 2 -> Inhibition de la synthèse d’eicosanoïdes pro-inflammatoires (prostaglandines, thromboxane, leucotriènes).
Inhibition de la NO synthase inductible -> Diminution de la production d’espèces radicalaires.
- Effets immunosuppresseurs :
Diminution de l’expression des molécules du CMH II -> Diminution de l’antigénicité des protéines.
Inhibition de la production d’IL-2 -> Diminution de la prolifération lymphocytaire.
- Effets pro-apoptotiques :
Induction de gènes de mort cellulaire ou répression de facteurs ou de gènes indispensables à la vie cellulaire -> Mort cellulaire.
Puissance et Efficacité Clinique
La puissance d'un GC est calculée comme l'inverse de sa CE50 (concentration produisant l'effet maximal de moitié).
Puissance relative anti-inflammatoire : La puissance relative des GCs varie selon le biomarqueur utilisé (e.g., suppression des lymphocytes T helper vs. suppression du cortisol).
Effets Minéralocorticoïdes : Les GC sont également classés par leur puissance minéralocorticoïde.
| Glucocorticoïde | Durée d’action | Effet minéralo-corticoïde | Puissance anti-inflammatoire | Equivalence de dose |
|---|---|---|---|---|
| hydrocortisone (= cortisol) | courte | 1 | 1 | 20 mg |
| prednisone | intermédiaire | 0.8 | 4 | 5 mg |
| prednisolone | intermédiaire | 0.8 | 4 | 5 mg |
| méthylprednisolone | intermédiaire | 0.5 | 5 | 4 mg |
| triamcinolone | intermédiaire | 0 | 5 | 4 mg |
| bétaméthasone | prolongée | 0 | 25 | 0,75 mg |
| dexaméthasone | prolongée | 0 | 25 | 0,75 mg |
Note : durée d’action courte : 8-12h, intermédiaire : 12-36h, prolongée 36-72h
Pharmacocinétique
Présentations
Les GC sont disponibles sous forme orale et parentérale mais également sous forme inhalée, topique, collyre, spray nasal, lavement, injectable (intra-articulaire, intra-musculaire, intra-auriculaire ou rachidienne).
Absorption et biodisponibilité
Selon la voie
- Voie orale : Bonne absorption digestive, mais biodisponibilité variable selon la molécule (cf. ci-dessous).
- Voie parentérale : Utilisée en urgence (ex. : méthylprednisolone IV), avec une biodisponibilité de 100 %.
- Voie inhalée/cutanée : Absorption systémique possible, surtout en cas d’application sur de grandes surfaces ou de muqueuses lésées.
Selon la molécule
- La prednisone (CORTANCYL®) est une prodrogue qui doit être convertie en prednisolone (forme active) par l’enzyme 11β-hydroxystéroïde déshydrogénase de type 1 (11β-HSD1) dans le foie.
Cette conversion est variable selon les individus en raison de :
- polymorphismes génétiques de l’enzyme 11β-HSD1, qui peuvent réduire ou accélérer la conversion ;
- fonction hépatique : en cas d’insuffisance hépatique, la conversion est moins efficace, ce qui réduit l’effet thérapeutique ;
- interactions médicamenteuses : certains médicaments (comme la rifampicine ou le phénobarbital) induisent le cytochrome P450 et peuvent accélérer le métabolisme de la prednisone, réduisant ainsi sa biodisponibilité.
- La prednisolone (SOLUPRED®) est déjà sous sa forme active et ne nécessite pas de conversion hépatique.
Cependant, sa biodisponibilité est plus variable (60 à 90%) car influencée par :
- absorption intestinale : Variable selon le pH gastrique, la motilité intestinale, ou la présence d’aliments ;
- métabolisme de premier passage : bien que la prednisolone soit active, une partie est métabolisée avant d’atteindre la circulation systémique, ce qui peut varier selon l’activité des enzymes hépatiques (comme le CYP3A4) ;
- variabilité génétique : les transporteurs intestinaux (comme la P-glycoprotéine) peuvent influencer son absorption.
Distribution et liaison protéique
Les corticoïdes circulent liés à des protéines plasmatiques (transcortine [globuline liant les corticostéroïdes] et albumine), seule la fraction libre (10–20 %) est active.
Particularités :
Prednisone et Prednisolone : pharmacocinétique non linéaire, car ils se lient à la transcortine avec une haute affinité mais une faible capacité, et à l'albumine avec une faible affinité mais une haute capacité. Lorsque la transcortine est saturée (à des doses supérieures à 20 mg pour l'hydrocortisone et la prednisolone), la fraction de GC libre augmente de manière non linéaire.
Méthylprednisolone et Dexaméthasone : aucune affinité pour la transcortine, liaison uniquement à l'albumine, d’où une pharmacocinétique linéaire.
Métabolisme et élimination
- Métabolisme hépatique : Les corticoïdes subissent une réduction, une hydroxylation, puis une conjugaison (glucuronidation, sulfatation), les rendant inactifs.
- Excrétion rénale : Principalement urinaire sous forme de métabolites inactifs.
NB : Demi-vie plasmatique vs biologique : La demi-vie plasmatique (ex. : 90 min pour le cortisol, 3–4 h pour la prednisone) ne reflète pas la durée d’action biologique, souvent plus longue (ex. : 18–36 h pour la prednisone) en raison de la persistance de l’effet génomique.
Interactions médicamenteuses pharmacocinétiques
Les GC sont des substrats du cytochrome P450 3A4.
Inducteurs enzymatiques du CYP3A4 (ex : phénobarbital, carbamazépine, rifampicine) : augmentation de la clairance, donc diminution de la demi-vie de la prednisolone et de la méthylprednisolone.
Inhibiteurs enzymatiques du CYP3A4 (ex : kétoconazole, clarithromycine, diltiazem) : diminution de la clairance, donc augmentation de la demi-vie de la méthylprednisolone et de la dexaméthasone. Prednisolone généralement non affectée.
Contraceptifs oraux (COs) : Les COs peuvent augmenter les concentrations plasmatiques et diminuer la clairance de la prednisolone (via l'augmentation des niveaux de transcortine) et de la méthylprednisolone (via l'inhibition des processus oxydatifs).
Effets Secondaires et Toxicité
Les effets indésirables des GC résultent de leurs effets pléiotropes et sont généralement associés à un excès d'activité glucocorticoïde. Le risque de survenue et la gravité de ces effets sont fortement liés à la dose et à la durée du traitement. Les effets indésirables les plus fréquents et les plus graves surviennent pour des expositions prolongées.
La physiopathologie des effets indésirables est intrinsèquement liée aux mécanismes d'action des GC, principalement lié à la transactivation (action transcriptionnelle directe).
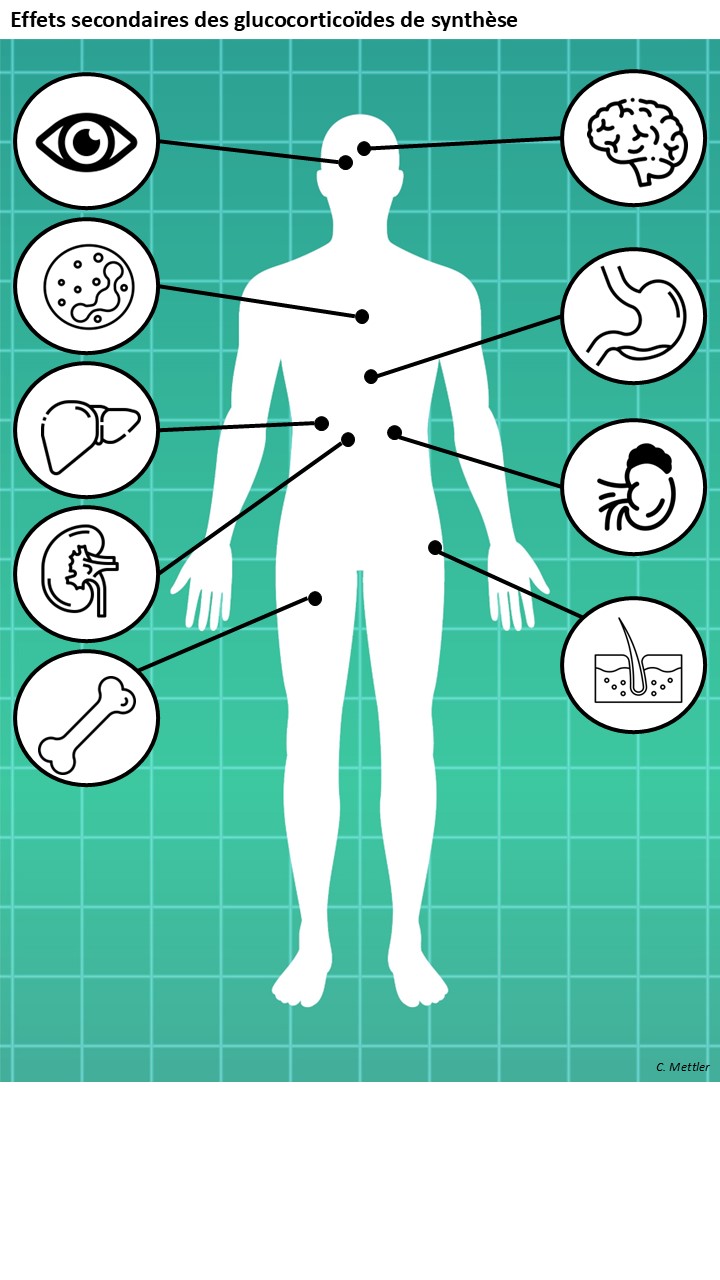
Figure [Effets secondaires des glucocorticoïdes de synthèse] interactive : pour chaque figure apparition d’un encart
 |
Effets neurologiques : changements dans le comportement, la cognition, la mémoire et l'humeur, trouble du sommeil |
 |
Suppression de l'axe hypothalamo-hypophyso-surrénalien Mécanismes : régulation négative sur l'axe hypothalamo-hypophyso-surrénalien -> inhibition de la sécrétion de cortisol par les glandes surrénales -> atrophie surrénalienne Conséquences : L'arrêt brutal du traitement peut provoquer des signes d' insuffisance surrénalienne aigue . La CE50 nécessaire pour la suppression du cortisol est inférieure à celle requise pour la suppression des lymphocytes T auxiliaires, ce qui souligne la difficulté à dissocier les effets thérapeutiques des effets secondaires endocriniens. |
 |
Effets digestifs : saignements gastro-intestinaux, ulcères digestifs, pancréatite |
 |
Effets cutanés Mécanismes : suppression de la prolifération des cellules cutanées et de la synthèse de protéines (comme le collagène et les métalloprotéinases matricielles). Conséquences : atrophie cutanée, ecchymoses, inhibition de la cicatrisation |
 |
Effets sur le système musculo-squelettique et la croissance Mécanismes : Réduction de la prolifération et induction d’apoptose des chondrocytes dans la plaque de croissance des os longs. Liaison aux éléments de réponse négative aux glucocorticoïdes (nGREs) -> inhibition de la transcription de l'ostéocalcine (protéine essentielle à la minéralisation osseuse) dans les ostéoblastes ; apoptose des ostéoblastes et augmentation de l'activité des ostéoclastes. Dégradation des protéines en acides aminés libres dans les muscles. Conséquences : retard de croissance chez l’enfant, ostéoporose chez l’adulte, ostéonécrose, amyotrophie |
 |
Effets cardiovasculaires et rénaux Mécanismes : effets minéralocorticoïdes -> rétention sodique, excrétion du potassium ; potentialisation des réponses vasopressives à l'angiotensine II (due à l'induction des récepteurs à l'angiotensine II). Conséquences : hypertension artérielle, rétention hydrosodée, hypokaliémie |
 |
Troubles métaboliques et endocriniens Mécanismes : activation de la gluconéogenèse, du catabolisme protéique et la mobilisation des acides gras. Conséquences : Diabète, dyslipidémie, obésité, syndrome de Cushing |
 |
Immunosuppressions Mécanismes : Inhibition des lymphocytes T (notamment Th1 et Th17), réduisant la réponse cellulaire contre les virus, bactéries intracellulaires et champignons. Diminution de la production de cytokines pro-inflammatoires (TNF-α, IL-1, IL-6), essentielles pour la défense contre les pathogènes. Altération de la fonction des macrophages et des neutrophiles , réduisant la phagocytose et la réponse innée. Augmentation de la perméabilité intestinale et pulmonaire, favorisant la translocation bactérienne (ex. : Pneumocystis jirovecii, Candida). Conséquences : augmentation du risque infectieux. - Infections bactériennes : Listériose (Listeria monocytogenes) : Risque multiplié par 2 à 3 chez les patients sous GC à haute dose (> 20 mg/j d’équivalent prednisone) ; - Tuberculose (Mycobacterium tuberculosis) : Risque accru de réactivation ou de progression d’une infection latente (> 15 mg/j pendant > 4 semaines) ; - Infections à Staphylococcus aureus. - Infections virales : Varicelle-zona (VZV) ; Herpès simplex (HSV) et cytomégalovirus (CMV). - Infections fongiques : Pneumocystose (Pneumocystis jirovecii) (> 16 mg/j pendant > 2 semaines) ; Candidose invasive ; Aspergillose. - Infections parasitaires : Anguilulose ou Strongyloïdose (Strongyloides stercoralis). |
 |
Effets oculaires : cataracte (sous-capsulaire postérieure), glaucome |
Perspectives
Le défi thérapeutique réside dans le développement de ligands glucocorticoïdes sélectifs (SEGRAS) qui pourraient induire la transrépression anti-inflammatoire tout en minimisant la transactivation responsable des effets secondaires, offrant ainsi un meilleur indice thérapeutique.
Les GC, grâce à leurs propriétés anti-inflammatoires et immunosuppressives puissantes, restent des outils thérapeutiques incontournables dans de nombreuses pathologies, qu’elles soient aiguës ou chroniques. Leur utilisation doit cependant être équilibrée et réfléchie car leur efficacité s’accompagne d’un cortège d’effets secondaires potentiellement graves, allant des troubles métaboliques aux complications infectieuses, en passant par des risques osseux et cardiovasculaires. Leur pharmacodynamie complexe et leur pharmacocinétique variable imposent une individualisation rigoureuse des schémas thérapeutiques, en tenant compte des spécificités du patient (âge, comorbidités, fonction hépatique, ...). La juste prescription repose ainsi sur un équilibre délicat : maximiser les bénéfices cliniques tout en minimisant les risques, grâce à une évaluation pré-thérapeutique approfondie, une surveillance régulière, et une adaptation posologique dynamique.
Dr Camille Mettler (médecine interne, Paris)